NNadir
NNadir's JournalThe Effect of Climate Change on Nuclear Reactor Cooling Will Be Studied at Argonne National Lab.
Argonne researching “climate-ready” nuclear plant design (Nuclear News, April 24, 2024.)Argonne will use funding from the Department of Energy’s Gateway for Accelerated Innovation in Nuclear to study climate-ready options for nuclear reactor designs. The goal is to develop a backup plan for nuclear cooling systems in the event that primary water sources are compromised by global warming—and Energy Northwest is partnering in the study to consider cooling options as river conditions may change near its Columbia nuclear power plant in Richland, Wash.
“It’s a very commendable way of thinking about climate change—to plan before doing something versus not thinking about it and trying to adapt afterwards,” Rao Kotamarthi, senior scientist in Argonne’s Environmental Science Division, said of Energy Northwest’s efforts. “A lot of people are confused about how to use the global climate data that exists, to make it actionable. At Argonne, we are working to provide very regional climate data in a form that industry can act on...”
...Cooling options: Vilim said local flowing water is the most economical and best way to cool a reactor, which is the current design of Washington’s nuclear plant. But changing climate models predict hotter, drier days in that region, which will affect the volume, flow, and temperature of the Columbia River.
“Dry cooling is not quite as efficient or as economical as wet cooling, but if wet cooling isn’t available, it’s your best option,” Vilim said.
Dry cooling uses ambient air circulated across a reactor’s heat exchangers instead of relying on a river or lake to conduct heat away from a reactor, using fans or physics similar to those in a house chimney or car radiator.
Recently I had an unpleasant conversation (is there any other kind?) with an antinuke who displayed phenomenal ignorance of nuclear technology (is there any other kind of antinuke other than those who display phenomenal ignorance of what they deign to criticize?) who pointed, albeit in a very, very stupid way, making up numbers as it went along, to the fact that nuclear plants, like many gas and coal plants, require cooling water.
It is true nuclear plants require cooling water, albeit nothing like the ignorant antinuke implied. This aspect is a function of the fact that the US nuclear fleet was largely built in the 7th and 8th decade of the 20th century, where Rankine devices were the "thing" for all thermal power plants, nuclear plants being a subset of thermal plants. All Rankine plants have low thermal efficiency, taken, as a rule of thumb, at around 33%.
Antinuke mysticism has left us with climate change, and in another case of arsonists complaining about forest fires, I suppose this reality, which is the worst (and only) major environmental effect associated with nuclear plants, will drive antinukes to help their fossil fuel friends by advocating for the closure of nuclear plants, rather than refurbishment.
The Biden administration, along with Governors like Gretchen Whitmer, is working to sustain and restore nuclear infrastructure. In terms of support for nuclear energy infrastructure - motivated by the best reason for doing so, climate change - the Biden administration is clearly the best in modern times. As a long time advocate of nuclear energy as our last best hope for addressing climate change - reactionary funding of so called "renewable energy" is a grotesque and extremely expensive failure at doing so - I am very pleased that the leader of our party has set aside the rote antinukism that has been our biggest policy stain, our most dubious position.
The absolute failure to address climate change by any means, can and will however, impact all thermal power plants, plants which overwhelmingly supply the nation's electricity, led in modern times by gas plants, followed by coal followed by nuclear. Although nuclear energy has been the most successful at ameliorating climate change of all technologies, accounting (as of this writing) more than a year's worth (35 billion tons) of the dangerous fossil fuel waste carbon dioxide being dumped by fossil fuel powered devices, it is vulnerable to climate change. Shutting nuclear infrastructure thus represents another potential feedback loop that must be arrested.
I'm glad to see one of our nation's premier national labs on the case.
It is my hope that many of the new designs in the new era of nuclear energy creativity that has returned to our country, will minimize the need for cooling by exergy recovery, that is putting the heat to use in what are called "heat exchanger networks" connected with the general approach known as "process intensification." This is an approach of recovery more of the energy of heat, simply efficiency improvements. I understand the new generation - the generation we screwed by my generation attacking nuclear energy while ignoring fossil fuels, as ass-backwards as you can get - is on the case; I note that young people represent the largest class of pronuclear activists.
Nevertheless, we have to preserve to the best of our ability our existing nuclear infrastructure, and it does seem that some retrofits will need to be designed to address the problem. We can no longer tolerate the German/New York/and other policy areas replacing clean nuclear energy with filthy and deadly fossil fuels. The Argonne effort is therefore worthy of applause.
Really 5000 "degrees," higher than the boiling point of zirconium metal?
The core at Fukushima vaporized? Who knew?
I note the switch from every nuclear reactor for this made up figure (with no units) of 5000 "degrees" to the big, big, big, big Bogeyman at Fukushima.
It would be interesting to learn of something called a reference from a reputable source for this extraordinary claim of 5000 "degrees" but I'm familiar, certainly, over all the years of antinukes cheering for the destruction of the planet by the application of fear and ignorance, with which only antivaxxers can compare in terms of death tolls, although Covid never killed roughly 19,000 people day like air pollution - not even counting climate change - does. In most cases antinukes don't have references. It's easier to make stuff up. However, if one relies on "making stuff up" one risks confronting people with legitimate real knowledge of the case.
Here is something called a reference, with textual commentary from a reputable source (the premier medical journal Lancet) for the number of people killed by air pollution egged on by antinukism:
Here is what it says about air pollution deaths in the 2019 Global Burden of Disease Survey, if one is too busy to open it oneself because one is too busy carrying on about Fukushima:
I keep this handy to point out how little antinukes care for stuff that matters, be it climate change, or the other effects of fossil fuel use, about which antinukes couldn't care less.
Here also, is something called a reference to the number of deaths caused by radiation at Fukushima, as opposed to the number of people killed by fear of radiation at the same event:
It's open sourced, but an excerpt is relevant:
I added the bold.
I also keep this text handy as well whenever people are carrying on insipidly about the big, big, big, big, big Bogeyman at Fukushima on their computers, powered by electricity that overwhelming comes from fossil fuels, with fossil fuel waste killing millions of people per year, roughly 80 to 90 million people since people began whining about Fukushima 13 years ago.
As for the commentary on how solar cells work, let me say this: I am used to contempt for science, in particular the laws of thermodynamics, among the "renewable energy will save us" crowd, although it's very clear after 50 years of such chanting, the world is not saved. Climate change is getting worse faster, and one reason is the trillions of dollars spent uselessly on solar and wind energy with the effect of entrenching the use of fossil fuels. It is antinuke fear and ignorance that has left the planet in flames. The evocation of wishful thinking about magical batteries is, as I often point out, an expression of contempt for the second law of thermodynamics, which is slightly arcane, but accessible to anyone of reasonable intelligence now that rational explanations exist, developed in the late 19th and early 20th centuries. It is unusual however, for antinukes, as badly educated as they generally are, to hold the first law of thermodynamics in contempt, as is happening in this exchange. It's a much simpler law.
As for nuclear energy:
Nuclear energy saves lives.
Here is something called a reference, coauthored by one of the world's leading (and famous) climate scientists, making the point, which I also keep handy:
Prevented Mortality and Greenhouse Gas Emissions from Historical and Projected Nuclear Power (Pushker A. Kharecha* and James E. Hansen Environ. Sci. Technol., 2013, 47 (9), pp 4889–4895)
The problem, as I see it, is a persistent refusal to have a simple sense of decency, for the elevation of dogma entrenching ignorance. I have seen here, among the antinukes, people citing the toxic idiot Helen Caldicott's unreferenced writings rather like right wing Christian Fundamentalists quoting the Book of Genesis in the Bible to explain the existence of the universe. One really doesn't want to believe that this sort of thing goes on, but it does.
No amount of information can change the rhetoric of a cult, not religious cults, not antinuke cults, not political cults like Trumpism, Nazism or the like. Cult thinking kills people, every time, all the time. The current case is no different.
Have a very pleasant Friday.
Meet General Shawn Harris, the Democrat who seeks to oust Marjorie Taylor Greene
Meet Shawn Harris, the Democrat who seeks to oust Marjorie Taylor GreeneHe connected with the Washington Blade last week to discuss his candidacy as a Democrat in Georgia’s deep-red 14th Congressional District — and why his promise to deliver for constituents who have been failed by their current representative is resonating with voters across the political spectrum...
... “I was military and I was high-level military,” he said. “So, it’s very easy for Republicans to Google my name and look at my history. My last assignment was in Israel. So that was a very high-level position.”
“The second piece,” Harris said, “is I raise cattle. I raise Red Angus cattle. I’m actually in my office looking out the window at them right now.”
He noted that agriculture dominates Georgia’s economy, particularly “cattle and wineries,” and also said he is an active member of the Georgia Cattlemen’s Association and U.S. Cattlemen’s Association.
“Most cattlemen, at least here in this area, are Republicans,” Harris said, so during the group’s meetings, “they get a chance to meet me just as Shawn, just as another cattleman.” At the same time, he said, “they come out here and visit me on the farm” and vice-versa...
I definitely hope this guy has a serious shot. That awful woman needs to be gone. She's a traitor, a fool, and an embarrassment to our country.
Formal "Justification" for a Fast Lead Cooled Nuclear Reactor Sought in the UK.
Justification sought for use of Newcleo reactor in the UKSubtitle:
Excerpts:
...The NIA noted that a justification decision is one of the required steps for the operation of a new nuclear technology in the UK, but it is not a permit or licence that allows a specific project to go ahead. "Instead, it is a generic decision based on a high-level evaluation of the potential benefits and detriments of the proposed new nuclear practice as a pre-cursor to future regulatory processes," it added...
..."Advanced reactors like Newcleo's lead-cooled fast reactor design have enormous potential to support the UK's energy security and net-zero transition, so we were delighted to apply for this decision," said NIA Chief Executive Tom Greatrex. "This is an opportunity for the UK government to demonstrate that it backs advanced nuclear technologies to support a robust clean power mix and to reinvigorate the UK's proud tradition of nuclear innovation. We look forward to engaging with the government and the public throughout this process and to further applications for new nuclear designs in the future..."
..."We continue to progress our UK plans at pace - aiming to deliver our first of a kind commercial reactor in the UK by 2033. We are but one player in the new nuclear renaissance and we look forward to working with government and the rest of the sector to develop the robust supply chain that can deliver the UK's ambition of 24 GW of nuclear power by 2050..."
24 GWe of nuclear power in Britain definitely falls into the category of "too little, too late." Hopefully the goals will morph into something more aggressive.
Things are pretty dire as I write. I routinely calculate from the data at Weekly average data for CO2 concentrations measured at Mauna Loa], setting the second derivative d2C/dt2 equal to the difference between the running 52 week average for the rate of CO2 at the current date (as of this writing 24.87 ppm/10 years) with that of the value in the first week of 2000 (15.36 ppm/10 years). I then integrate twice and substitute the boundary conditions, the concentration of CO2 reported this week, and the current running average for 10 year increases to obtain a quadratic. If the fossil fuel indulgent antinuke rhetoric continues to triumph, the resulting quadratic (as of this week) works out to a concentration of around 519 ppm "by 2050." I expect that 24 GW of additional nuclear power in Britain would barely cover the growth in electricity demand, especially if the very dubious "electrify everything" scheme is allowed to proceed on the dubious basis driven by the difficult to break but very dangerous belief that so called "renewable energy" will be something other than useless, which won't happen.
Britain, I should note, has the world's largest supply of isolated reactor grade plutonium, and thus a lead cooled fast reactor would allow for the use of this plutonium to create more plutonium which will certainly be needed if there is to remain any slim hope of avoiding even worse climate change than we are already experiencing as the result of antinukism's grand propaganda success that has left the planet in flames.
Lead coolants are very attractive to my mind for a number of reasons connected with process intensification - providing high temperatures to meet a number of missions not merely connected to producing electricity. Historically, these types of reactors have actually operated on LBE, lead bismuth eutectics, but I note that the use of pure lead will lead to the transmutation of some lead into the far less toxic element bismuth (an ingredient in the OTC product "Pepto-Bismol" ), thus generating LBE in situ. This would involve certain technical challenges, but I believe they might well be possible to address.
Have a pleasant work week.
An Interesting Experiment in Trying to Involve Journalists in Reporting Real Science Correctly.
One of the standard half-serious jokes I often repeat in my writings here when confronting articles from journalists linked and discussed here, typically about energy and the environment which are often sensationist and misleading, is that "One cannot get a degree in journalism if one has passed a college level science course with a grade of C or better."
Journalism, as is the case in the American political disaster in which journalists have been and are actively working to normalize an intellectually deficient and declining criminal fraud as a Presidential candidate, while demeaning as "too old" an activist highly functional President, is generally horrible where scientific issues are involved. One result of this marketing of bizarre ideology by journalists is climate change. I regard the major issue in climate change to be selective attention, for example, the magnification of the views of extreme scientific minorities in climate denial, or the demonization of nuclear energy, which I continue to insist is the last, best hope for humanity.
Thus it was with some interest that I came across this paper in a scientific journal in which scientists involved themselves with, and reviewed, journalism in connection with a very serious issue, PFAS (Per/Poly Fluorinated Alkylated Substances) contamination in Europe: PFAS Contamination in Europe: Generating Knowledge and Mapping Known and Likely Contamination with “Expert-Reviewed” Journalism Alissa Cordner, Phil Brown, Ian T. Cousins, Martin Scheringer, Luc Martinon, Gary Dagorn, Raphaëlle Aubert, Leana Hosea, Rachel Salvidge, Catharina Felke, Nadja Tausche, Daniel Drepper, Gianluca Liva, Ana Tudela, Antonio Delgado, Derrick Salvatore, Sarah Pilz, and Stéphane Horel Environmental Science & Technology 2024 58 (15), 6616-6627
Unfortunately I will not have much time to discuss this paper in much detail, but a few excerpts and a graphic follow.
From the introduction:
PFAS have been broadly detected in environmental media including surface water, groundwater, raw and finished drinking water, soil, air, landfill leachate, sewage sludge, food, and dust. (12?15) As examples, a recent analysis of tap water samples in the United States collected between 2016 and 2021 detected PFAS at 45% of tested locations, (16) and the 2019 French National Biomonitoring Programme measured serum levels of 17 PFAS and detected PFAS in 100% of the 993 participants. (17) It has been argued that PFAS have exceeded a “planetary boundary” of “widespread and poorly reversible risks associated even with low-level PFAS exposures” since global rainwater samples have PFAS levels above proposed regulatory limits designed to protect public health... (1)
...In addition to data gaps due to a lack of testing, the present study was motivated by two additional data gaps. First, unlike in the US where many PFAS testing datasets have been made public by federal and state governments, academic research groups, and environmental advocacy organizations, (19?22) there were very few publicly available data on PFAS contamination across the EU. Second, in most cases, legal protections for industry trade secrets and confidential business information prevent the public from knowing where PFAS are used and emitted. (23,24) An exception to this is the US Toxics Release Inventory program, which has required reporting since 2020 by some firms in a subset of industries for a small number of PFAS. (25) But in most cases, even if a specific company is known to produce and/or use PFAS, it is difficult or impossible for the public to know whether PFAS are used at individual facilities...
The authors enlisted five investigative journalists in five countries, Belgium, France, Germany, Italy, and The Netherlands, in the "FPP" or "Forever Pollution Project" and paired them with a team of PFAS scientists, noting " journalistic projects aiming at generating data in collaboration with scientists rather than reporting on already-produced data are not common."
Not common, indeed. I would expect that most scientists feel as I do, that science journalism (except generally when it appears in major science journals) that journalism is oblivious when reporting science.
Further down in the article:
The map which is interactive in the full paper, and, I think, at the link given in the caption but cannot be so here:
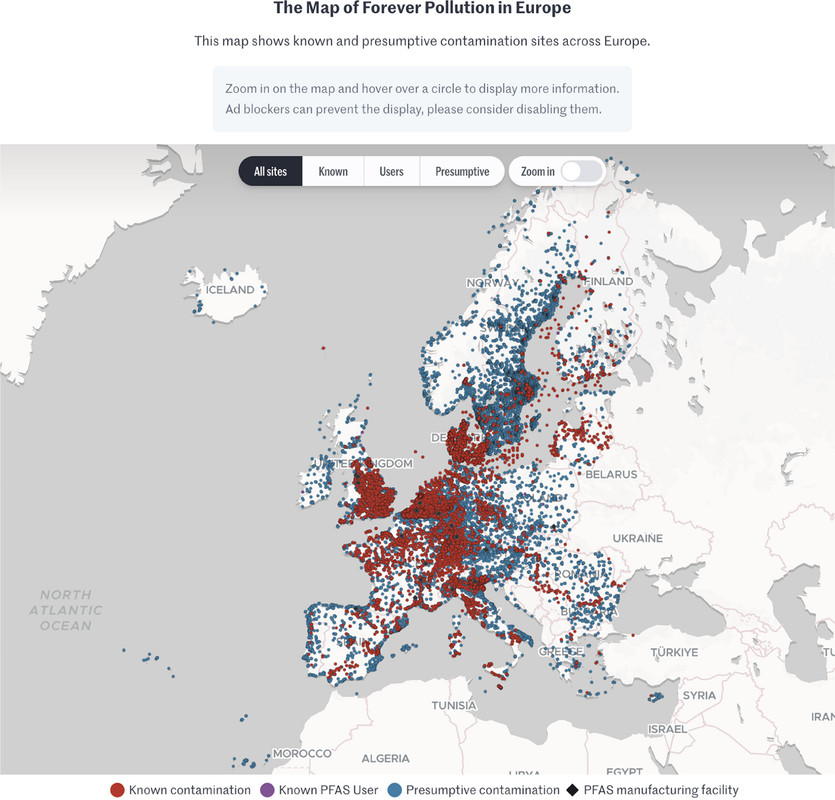
The caption:
I don't expect journalism to become a serious mirror of reality in my now short remaining lifetime, but we have to start somewhere; we have to try something, and I applaud this effort.
I wrote earlier today about PFAS and the very dangerous "hydrogen will save us" fantasy often hyped in the scientifically illiterate media earlier today:
A Brief Note on the Toxicology Associated With Hydrogen Fuel Cells.
Have a nice Sunday afternoon.
A Brief Note on the Toxicology Associated With Hydrogen Fuel Cells.
Intellectually, and morally, in my view, the hydrogen fuel fantasy should have been, and should be now, a non-starter, simply because of the laws of thermodynamics, notably the 2nd law. Like other "bait and switch" fantasies connected with diversion from the facts that fossil fuels can be rendered sustainable (or eliminated by wishful thinking) - including but not limited to sequestration, wind, and solar - the use of hydrogen fuels will make things worse, not better.
I laid out, in some detail, the facts connected with the nature of hydrogen as a front for the fossil fuel industry in a rather long post here: A Giant Climate Lie: When they're selling hydrogen, what they're really selling is fossil fuels.
This is also laid out in the scientific literature in the case of China, to which fossil fuel salespeople and salesbots who write here and elsewhere often appeal here to rebrand fossil fuels as "hydrogen:"
Subsidizing Grid-Based Electrolytic Hydrogen Will Increase Greenhouse Gas Emissions in Coal Dominated Power Systems Liqun Peng, Yang Guo, Shangwei Liu, Gang He, and Denise L. Mauzerall Environmental Science & Technology 2024 58 (12), 5187-5195.
One of the "bait and switch" tactics used to greenwash fossil fuels by talking about hydrogen is to talk about hydrogen fuel cells for cars. Hydrogen fuel cells actually are commercial products occasionally used for back up power for things like cell phone towers, although they are hardly mainstream.
One of the major environmental risks garnering increasing attention, is the issue of PFAS - "per (and poly) fluorinated alkylate substances, so called "forever chemicals" - which are known to have profound toxicological profiles. I know, but I don't believe that most people know, that the issue of PFAS is very much connected with hydrogen fuel cells, and that the widespread use of these, for instance in cars, would make this already intractable problem far worse.
A paper on strategies to degrade these has appeared in the scientific literature in an open source paper that I came across in my general reading. It is here:
Oxidative Transformation of Nafion-Related Fluorinated Ether Sulfonates: Comparison with Legacy PFAS Structures and Opportunities of Acidic Persulfate Digestion for PFAS Precursor Analysis Zekun Liu, Bosen Jin, Dandan Rao, Michael J. Bentel, Tianchi Liu, Jinyu Gao, Yujie Men, and Jinyong Liu Environmental Science & Technology 2024 58 (14), 6415-6424
Again, the paper is open sourced, and will be generally arcane for people not familiar with scientific punctilios, but a relevant paragraph referring to fuel cells and the relationship to PFAS is this one:
Nafion, which is used in all commercial fuel cells, is the a fluoropolymer of PFAS compounds.
Two figures from the paper:
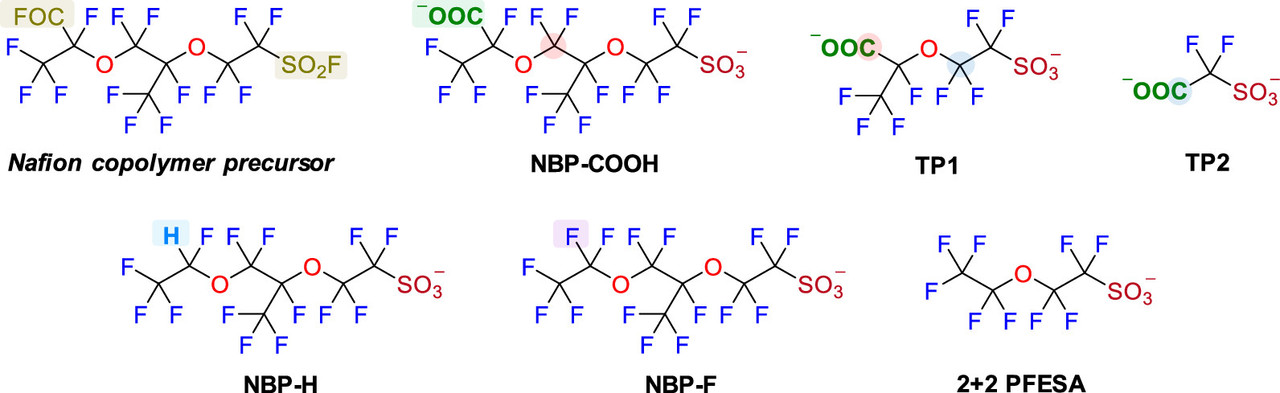
The caption:
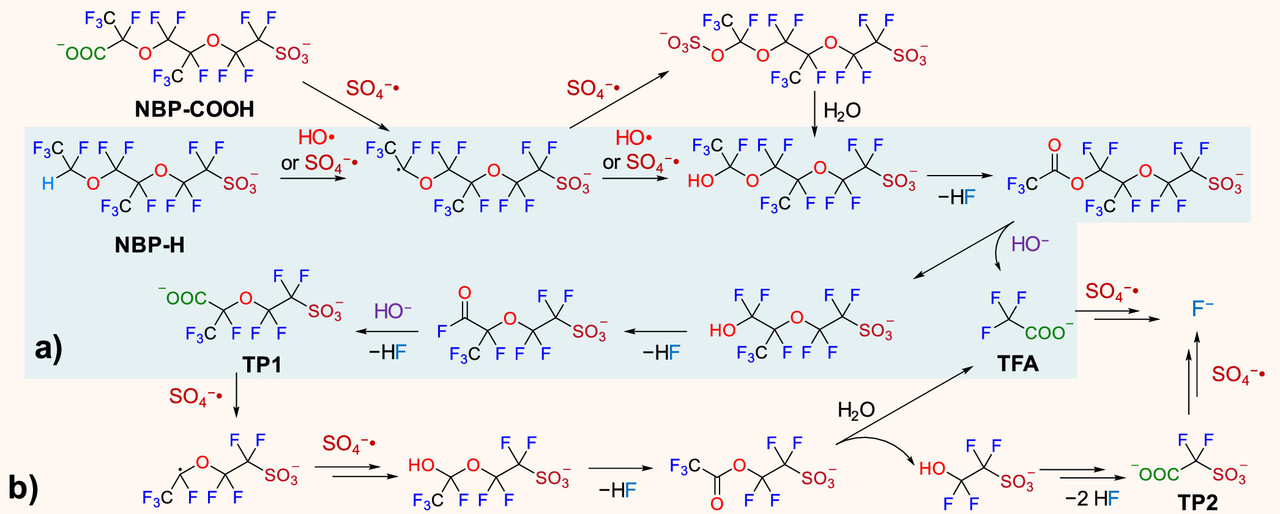
The sulfate and hydroxide radicals in this pathway require energy to create. I regard oxidative radicals of this type as the best pathway for mineralizing (destroying) PFAS. The type of energy ideal for this purpose is ionizing radiation, high energy radiation in the UV to gamma range. The energy for doing this is best obtained from radioactive materials, the easiest access to which can be obtained from used nuclear fuels.
Have a pleasant Sunday.
That scene in "My Dinner With Andre" About a Sexy Picture, and Looking at Pictures of My Wife When She Was Young.
Last week I wrote how my wife was going through a box of her late mother's pictures, and I found myself remarking on how beautiful she looked in a picture someone took of us at the North Rim of the Grand Canyon, just before we married, a picture that somehow ended up in my mother's-in-law collection and wasn't in ours.
My wife finally got around to going through her late Mom's photo albums.
A strange feature of the picture is that in it she actually looks younger than she was at the time; she was in her early twenties but looked like a teenager. By contrast, although I had hair and it was dark, and in the picture relatively long and wild in what may have been a wind, I looked older than I actually was; I was in my early thirties. (Because we looked as if the distance in our ages appeared to be larger than it was - my wife got "proofed" in restaurants and bars into her thirties - I used to get some dirty looks from time to time, as if I were an Epsteinish/Trumpish/Gaetzish sort of pervert.)
Well, in any case, I scanned the picture, and I've looked at it a number of times since doing so over the last week, fascinated by it. As I was doing so this morning, it suddenly struck me, looking at that picture of myself, that the man in the picture, me, didn't at that time know very much at all about the world; I was ignorant, even more arrogant than I am now, kind of smug, and although I was merely formally educated, but largely unacquainted with the deeper reality of the things to which my education merely exposed me.
Then I looked at my wife in the picture, and thought about who she was at the time, the Mona Lisa type smile she had on her face concealing the fact that she'd just emerged from a very unhappy upbringing and was possessed of a deep insecurity, and uncertainly - wisely I think - unsure whether it was actually a good idea to be with me, there and then.
In that beautiful movie My Dinner With Andre there's a scene where Andre Gregory discusses a picture of his wife, Mercedes "Chiquita" Gregory. From the script:
Script, My Dinner With Andre
The first time I went away with my (then future) wife, I took her to what was my favorite place in the universe, to Big Sur. We stayed in this beautiful small cabin in a grove of redwoods, a night I have never forgotten, and about which I often muse. I consider it the best night of my somewhat pathetic life up to that point - although in the years of marriage, many similarly beautiful moments would happily entail over and over again.
In the morning after that night in the cabin, we went on a hike up a small mountain in Pfeiffer Big Sur State Park, and we came upon a grove of pin oaks, and I took pictures of her up there in that grove.
There is one of those pictures in particular that has thrilled me ever since I took it and developed it almost 40 years ago, because in the picture, my wife has exactly the same expression she was wearing the very first time I saw her, the same pose, her legs stretched out, looking down almost shyly at her feet, detached from the outer world, but seemingly deep in an inner world, a kind of impenetrable look of something that could, should perhaps, be recognized as having an aura of buried pain, a resigned sadness strangely contrasting with a sublime physicality. In the Big Sur picture as opposed to my memory of first seeing her, she is sitting on a low branch of a massive oak the Big Sur grove, naked. I must have called that picture up a thousand times, but today, I find myself asking myself if I ever really saw it, I mean, saw it for real.
Of course, when I saw my wife for the first time, I was just another of the many puerile men possessed of less than noble erotic fantasies about her looks; I had no idea who she actually was. Of course, ultimately I did get to know her, and see her divorced from any kind of vanity, somewhat distressed about the somewhat dangerous situations into which her looks had gotten her although she managed to emerge largely unscathed. (Of course early on in our friendship, she could of suspected me of being another dangerous man of the type, but happily she didn't translate those things into suspicions of me or my intentions, although she might well have done so.)
Away from all that, the attentions of puerile men including me, she was always warm, kind, generous, funny and bright, surprisingly accessible and down to Earth, although the deeper things were kept to herself. I could not help falling in love.
But the point is, that in my admiration for these pictures of her youth, like Andre Gregory confessed in his own case, it's not act of seeing, but definitely an act of not seeing anything at all.
We were younger, better looking then, but frankly and honestly, as life winds down, I prefer who we are now. We hadn't lived at all then. We didn't know anything then. We've lived now. We know something, perhaps not a lot, but something now.
Most importantly, something we know now is what it is to have lived at all. That matters.
BWXT announces nuclear manufacturing plant expansion
BWXT announces nuclear manufacturing plant expansionAn excerpt:
BWXT is headquartered in Virginia and has 14 operating sites in the United States, Canada, and the United Kingdom. Its joint ventures provide management and operations at a dozen U.S. Department of Energy and NASA facilities.
What they are saying: “Our expansion comes at a time when we’re supporting our customers in the successful execution of some of the largest clean nuclear energy projects in the world,” said John MacQuarrie, president of commercial operations for BWXT. “The global nuclear industry is increasingly being called upon to mitigate the impacts of climate change and increase energy security and independence.
Premier of Ontario Doug Ford said, “We’re thrilled to see BWXT expand its footprint and create hundreds of new jobs in Cambridge. As our province continues to lead the future of nuclear energy, the company’s investment will help provide Ontario families and businesses with access to clean, reliable, and affordable electricity for generations to come...”
I added the bold.
This won't go over well in the cults that drive the so called "renewable energy"/fossil fuel nexus, but for humanity as a whole this is a good thing, building back better the nuclear manufacturing infrastructure destroyed by fear and ignorance, thus leaving the planet in flames.
Nuclear systems can do more than provide families "with access to clean, reliable, and affordable electricity for generations to come," although there's certainly nothing wrong with that. It's nice to see people thinking about future generations, something certainly ignored by advocates of the so called "renewable energy"/fossil fuel nexus, where future generations are treated with contempt.
Personally, though, I'm opposed on thermodynamic grounds, to the "electrify everything" bad idea that's gaining unwarranted popularity, but we have to restart somewhere. (Electricity is by its nature, thermodynamically degraded.) With the planet already in flames, notably with Canada for one subset of the climate burned regions, this comes under the rubric of "too little, too late" and is hardly enough, but the world seems to be overcoming the triumph of antiscience antinukism to try to save what is left to be saved and perhaps, even restore some of what can be restored.
Ontario is rising as a center of going nuclear against climate change, something of which citizens of the Province should be proud.
Non Nuclear Portions of Wyoming's Kemmerer Nuclear Power Plant to Start Construction This Summer.
TerraPower submits Natrium construction application to the NRCExcerpts:
TerraPower purchased land in Wyoming near one of the state’s retiring coal plants to deploy its Natrium sodium fast reactor technology. Kemmerer Power Station Unit 1 would operate a 345-MW sodium-cooled reactor in conjunction with molten salt–based energy storage...
...A closer look: Natrium will use liquid sodium as a coolant instead of water. According to TerraPower, the reactor features improved fuel utilization, enhanced safety features, and a streamlined plant layout that will require fewer overall materials to construct. Because of the reactor’s storage technology, it can boost the system’s output to 500 MWe for more than five and a half hours when needed to meet additional grid demand...
... Support for the project: Natrium is one of two advanced reactor demonstration projects selected for competitive funding through the Department of Energy’s Advanced Reactor Demonstration Program. (X-energy’s Xe-100 is the other.) TerraPower received $1.6 billion in funding from the Bipartisan Infrastructure Law signed by President Biden in November 2022, which is to be used to ensure the completion of the plant. The company has also raised more than $1 billion in private funding.
Last month, TerraPower announced the second round of contracts for long-lead suppliers supporting the development of the Natrium reactor.
I'm not a sodium coolant kind of guy myself, but this reactor has features of the "breed and burn" concept which will allow it ultimately to run on depleted uranium, if I understand the design well.
Light a Candle, an Innovative Burn Up Strategy for Nuclear Reactors. (Hiroshi Sekimoto)
This will help to generate plutonium necessary if we are to have any hope of addressing climate change.
Dr. Kathryn Huff Is Leaving DOE, Praising the Administration as She Goes.
After serving her country for three years, Dr. Kathryn Huff is leaving DOE:
Kathryn Huff stepping down from DOE Nuclear Energy post
Excerpt:
“Serving in this capacity has been an unparalleled privilege, and I’m immensely grateful for the opportunity to have worked alongside you--the dedicated and talented public servants in Nuclear Energy, in DOE, and across the Biden-Harris Administration,” Huff wrote in an email announcement to colleagues last week. “I chose this timing to enable the smoothest transition back to my professorship at the University of Illinois at Urbana-Champaign where my beloved research, students, husband, and dog await...”
... “Reflecting on the past three years, I’m astonished by the tangible progress the U.S. has made in nuclear energy,” Huff said in her note to colleagues. “Reactors once destined to shut down now have a role in our 2035 and 2050 goals, new reactors are coming online, and the commercialization of advanced reactors has begun. We’re also securing the front end of the nuclear fuel cycle, restarting a consent-based approach to spent nuclear fuel management, and expanding international cooperation on peaceful nuclear technology. Perhaps most notably, the U.S. joined a groundswell of other nations in committing to triple nuclear energy capacity by 2050 (to) address the climate crisis and improve energy security.”
She added, “I will, of course, continue to contribute to the advancement of nuclear energy however I can, for as long as I can, from wherever I am.”
I understand her reasons for leaving, but I'm a little sad to see her go. She was, to my mind, a star in the Biden-Harris administration, a high powered academic scientist bringing high level expertise to government. It does seem her accomplishments in the move building back better our nuclear power infrastructure have been significant, the most significant since Steven Chu, in the Biden administration started us on the now nearly complete path to the Vogel reactors.
Profile Information
Gender: MaleCurrent location: New Jersey
Member since: 2002
Number of posts: 33,518