NNadir
NNadir's JournalA comprehensive quantification of global nitrous oxide sources and sinks
The paper I'll discuss in this post is this one: A comprehensive quantification of global nitrous oxide sources and sinks (Tian, H., Xu, R., Canadell, J.G. et al. Nature 586, 248–256 (2020))
Nitrous oxide, N2O, laughing gas, has always been a part of the nitrogen cycle, but with the invention of the Haber-Bosch process in the early 20th century, on which our modern food supply depends, the equilibrium has been severely disturbed. Since, like CFC's, nitrous oxide is both a greenhouse gas and an ozone depletion agent. I'm sure I've written in this space and elsewhere about this serious environmental issue, since it worries me quite a bit. So this paper in Nature caught my eye. The authors are a consortium of scientists from around the world, who have assembled what may be the most detailed account of accumulations of this gas in the atmosphere.
The abstract is open sourced, and may be found at the link in the paper. It is worth pointing out the cogent portions of it however:
From the introduction to the paper:
The authors refer to "bottom up" analyses, which consists basically of multiple source terms such as known emissions, modeling of agricultural inputs based on field measurements, etc., and "top down" analyses which consist of measurements coupled to transport flux modeling. The utilize 43 N2O "flux estimates" 30 of which are "bottom up," 5 of which are "top down" and 8 models.
They write:
This cartoon figure gives a feel for sources:
Fig. 1: Global N2O budget for 2007–2016.
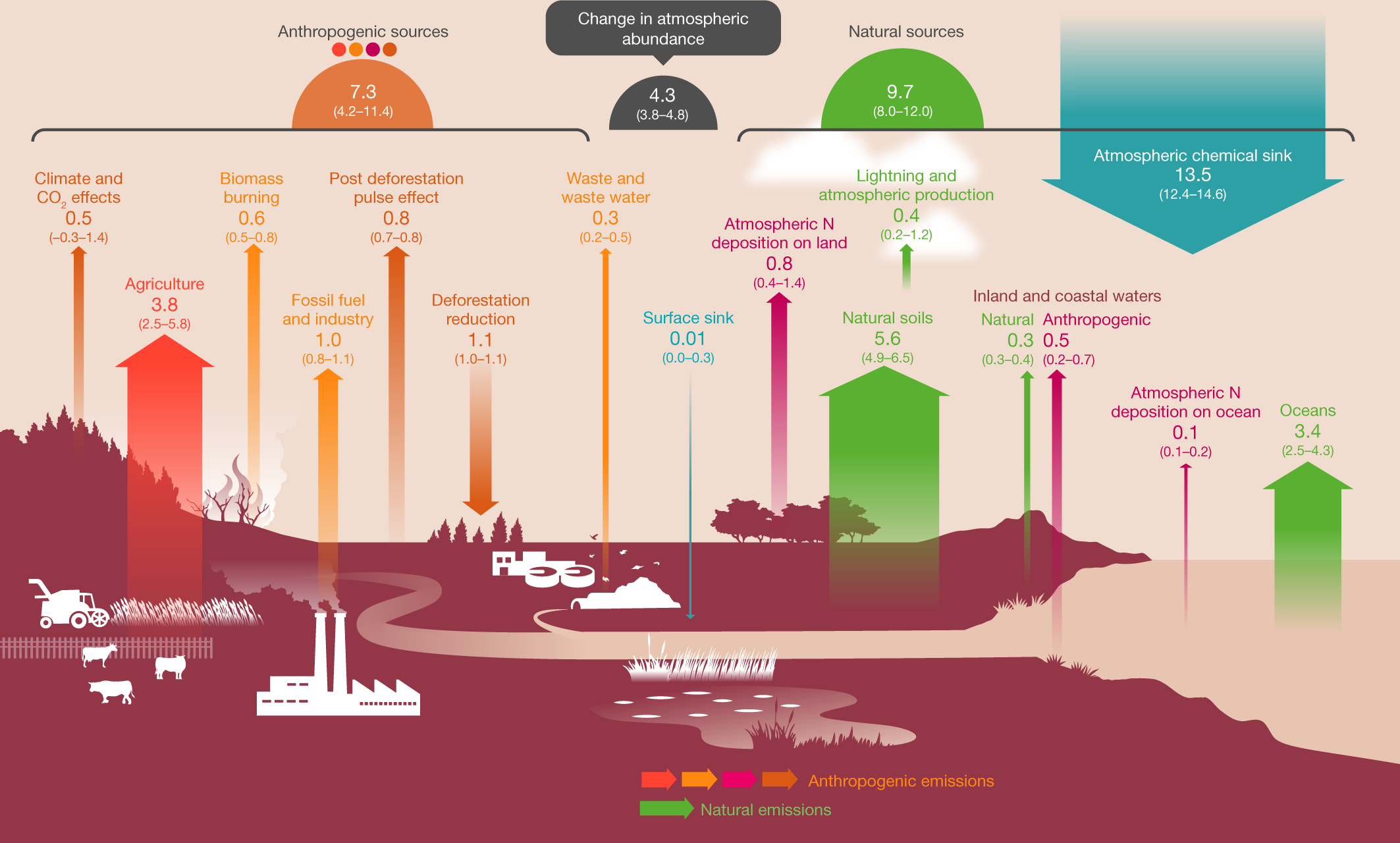
The caption:
A Tg - terragram - is, of course, a million metric tons.
Isotopic analyses can show the sources, by the way, of N2O. For example, N2O in the troposphere is slightly enriched in the O17 isotope, which is believed to be connected with interactions of atmospheric ammonia and the dangerous fossil fuel combustion waste NO2. The origin of the anomalous or “mass‐independent” oxygen isotope fractionation in tropospheric N2O (Crutzen, et al., Geophys. Res. Lett. Volume 28, Issue 3 1 February 2001 Pages 503-506)
In the paper under discussion there is a rather large table l will not reproduce here that breaks down various source inputs and outputs, natural and anthropogenic. The mean value for concentrations of nitrous oxide in the atmosphere have risen from a mean value of 1,462 Tg, 1.462 billion tons, to a mean value in the period between 2007 and 2016 of 1,555 Tg, 1.555 billion tons, an increase of 93 million tons.
A breakdown of inputs represented graphically:
Fig. 2: Regional N2O sources in the decade 2007–2016.
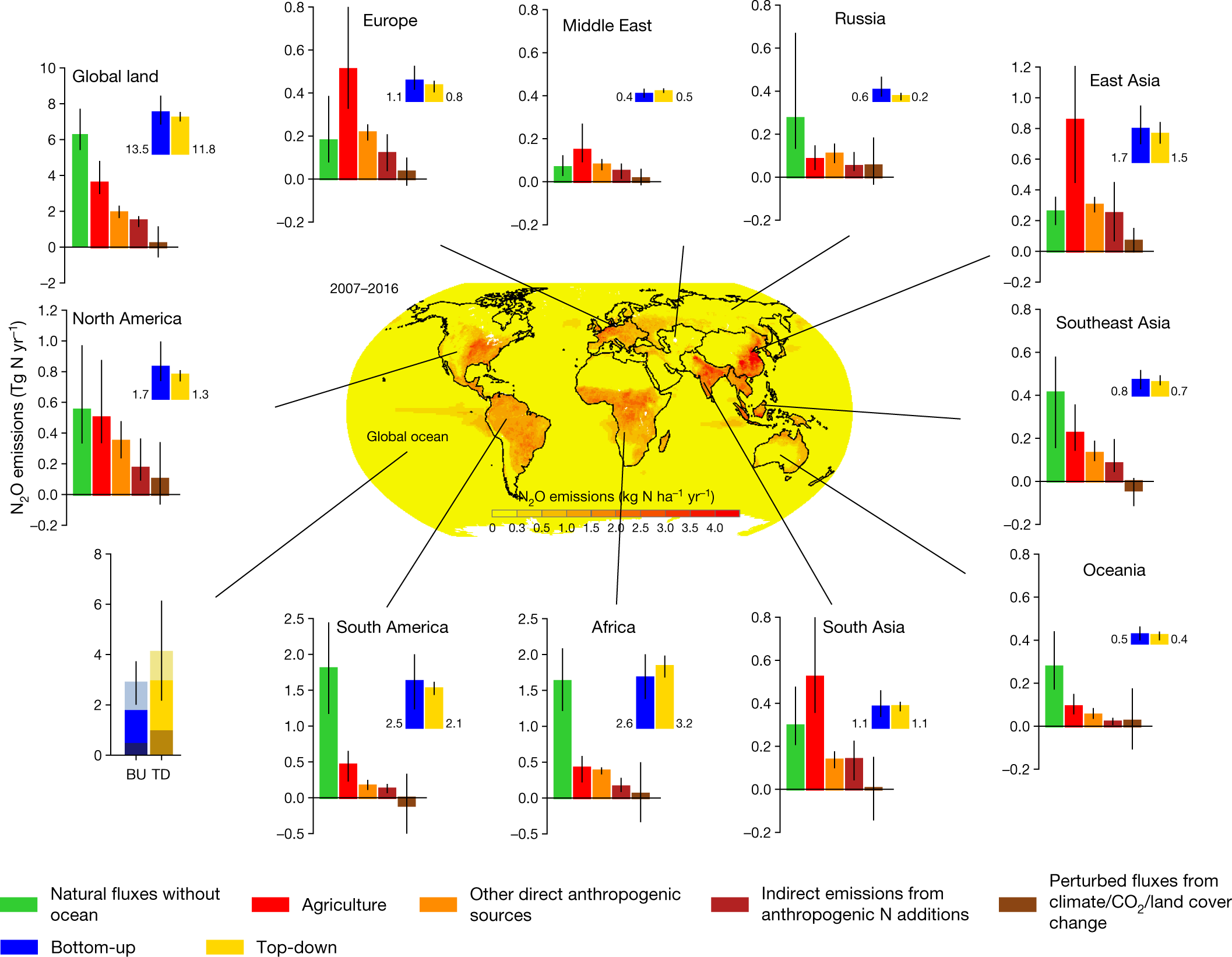
The caption:
Fig. 3: Ensembles of regional anthropogenic N2O emissions over the period 1980–2016.
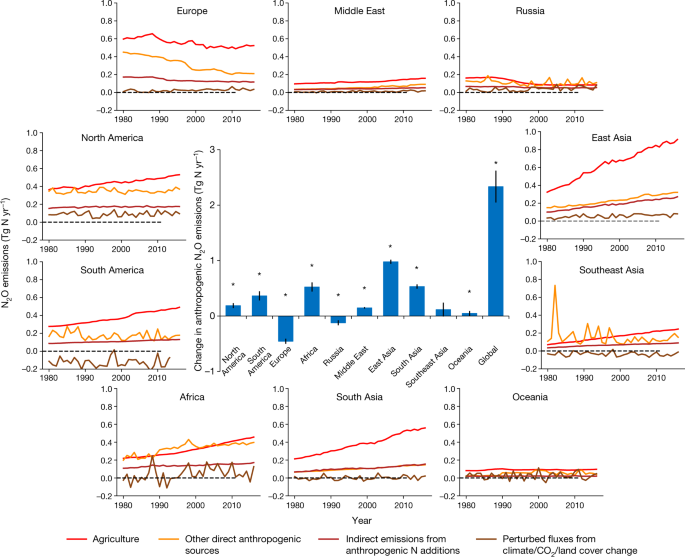
The caption:
Predictions:
Fig. 4: Historical and projected global anthropogenic N2O emissions and concentrations.
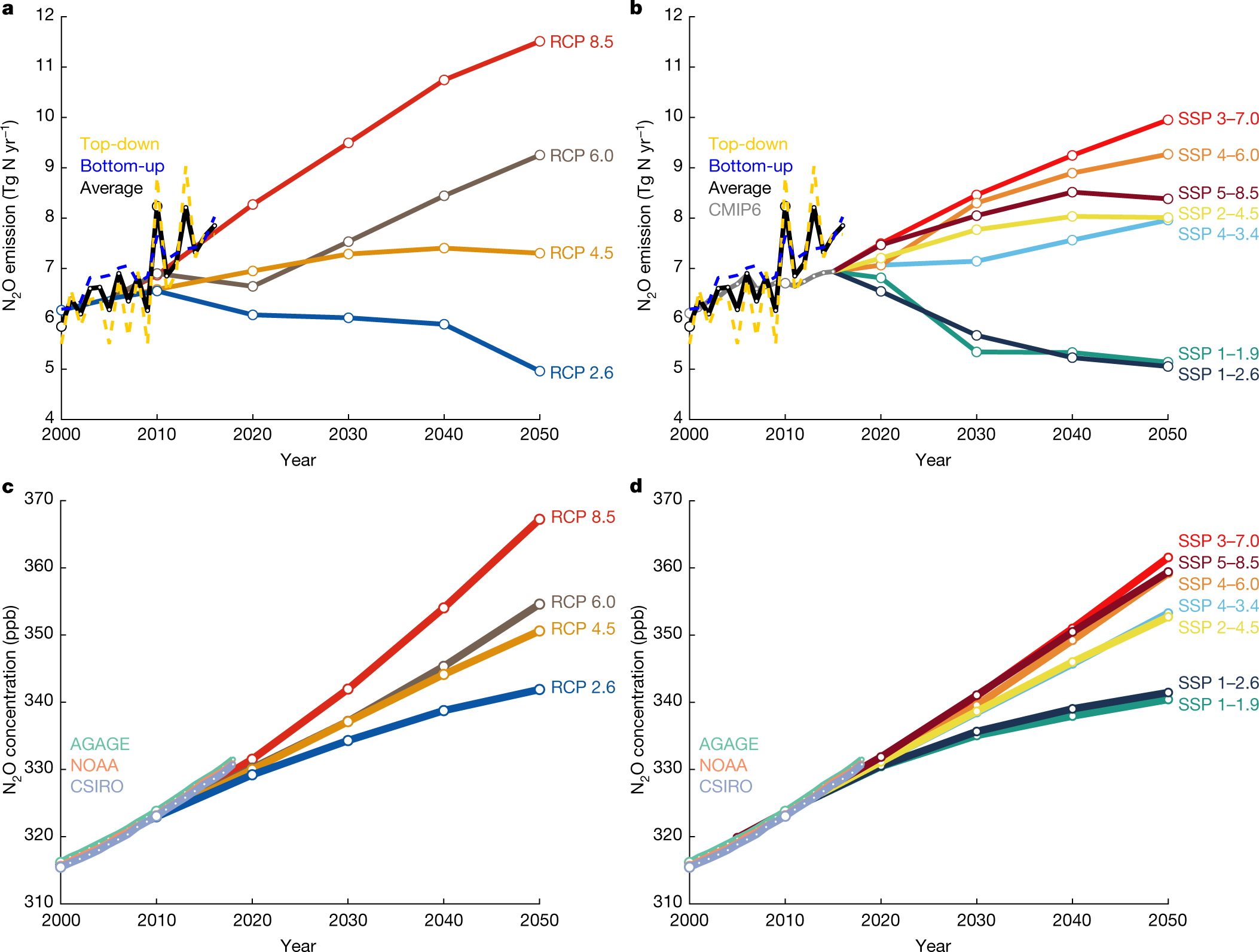
These IPCC reports by the way, are bitterly comical in all of their optimistic scenarios, "RCP" Representative Concentration Pathways. Things are not even "business as usual" as described in the early editions. Things are worse than business as usual.
This is true here. The authors write:
Nitrous oxide will support combustion; it is an oxidizing gas. In theory it would be reduced by fire, but in general, combustion also generates significant higher nitrates, NO and NO2 which are ultimately transformed into N2O.
The main sink is photolytic, at wavelengths below 230 nm, which is in the UV range. Singlet oxygen, a high energy state of oxygen gas, also can destroy N2O. It is also generated by radiation, as well as some chemical means.
In recent years, I have come to think of air as a working fluid in Brayton cycle/combined cycle power plants. I've returned to an old idea about which I thought much in the 1990s and early 2000's - although they were not directed at that time to Brayton cycles, but rather as Rankine cycles, which is to use highly radioactive materials as heat transfer agents, in those days, salts in the liquid phase, in these days salts and metals in the vapor phase. Under these circumstances, new sinks for highly stable or highly problematic greenhouse and ozone depleting agents, as well as particulates, might represent a new sink for not just nitrous oxide, but others.
I trust you are enjoying a safe, productive, and enjoyable weekend.
Electrochemical oxidation of 243Am(III) in nitric acid by a terpyridyl-derivatized electrode.
The paper I'll discuss in this post came out about 5 years ago, but somehow I missed it. The paper is: Electrochemical oxidation of 243Am(III) in nitric acid by a terpyridyl-derivatized electrode (Christopher J. Dares1, Alexander M. Lapides1, Bruce J. Mincher2, Thomas J. Meyer1,* Science Vol. 350, Issue 6261, pp. 652-655).
I've written several times about the element amercium, in this space, most recently about its separation from its f-series cogener, europium.
Liquid/Liquid Extraction Kinetics for the separation of Americium and Europium. The process described therein is a solvent based process, but utilizes specialized membranes. As I noted at that time, there are several drawbacks to the process.
However americium is potentially valuable fuel, particularly because of its nonproliferation value, since burning it will generate the heat generating isotope plutonium 238, (via the decay of Cm-242, formed from the decay of Am-242) which when added to other plutonium isotopes, can make them all useless in nuclear weapons.
It turns out, on further inspection, that Am-242 and its nuclear isomer Am-242m - both of which are highly fissionable - are among the best possible breeder fuels, having a high neutron multiplicity, meaning they can serve to transmute uranium into plutonium, greatly expanding the immediate availability of nuclear energy while avoiding isotope separations.
Here is the plot of the neutron multiplicity for the fission of Am-242(m):

Evaluated Nuclear Data File (ENDF) Retrieval & Plotting ("Neutron induced nubar." )
This spectrum, which is from the ENDF nuclear data files is clearly not highly resolved, to be sure, and surely represents a kind of average. It is undoubtedly difficult to obtain highly purified Am-242m for experimental verification. An interesting approach to obtaining it in high isotopic has been proposed, owing to its possible utility in making very small nuclear reactors for medical or space applications: Detailed Design of 242mAm Breeding in Pressurized Water Reactors (Leonid Golyand, Yigal Ronen & Eugene Shwageraus, Nuclear Science and Engineering, 168:1, 23-36 (2011)). To my knowledge, however, this proposal has never been reduced to practice.
There are very few nuclei that exhibit this high a multiplicity, over 3 neutrons per fission at any incident neutron energy. To my immediate knowledge, the only such nuclei is plutonium-241, and this over a fairly narrow range of incident neutron energies in the epithermal region.
I also discussed the critical masses of americium isotopes in this space: Critical Masses of the Three Accessible Americium Isotopes. In a fast nuclear spectrum, americium-241, the common americium isotope produced in the common thermal nuclear reactors that dominate the commercial nuclear industry, particularly in used fuel that has been stored for decades without reprocessing, is fissionable and thus is a potential nuclear fuel in its own right.
A "refinement" of some nuclear physics parameters relevant to the use of americium as a nuclear fuel was published about 13 years ago as of this writing, by scientists at Los Alamos: Improved Evaluations of Neutron-Induced Reactions on Americium Isotopes (P. Talou,*† T. Kawano, and P. G. Young, Nuclear Science and Engineering 155, 84–95 2007).
In a fast nuclear reactor, neutrons have an energy of 1 to 2 MeV whereas in a thermal reactor, the neutron speed is taken to be the kinetic energy of molecules in air, generally taken as 0.0253 eV, almost 100 million times lower energy. Thermalized neutrons tend to be absorbed without fission in americium-241, whereas at higher energy, they tend to fission. The following graphics from this paper show that for a region of neutrons well above the thermal region, but in the general region with the fission to capture ratio climbs:


The next graphic, from the same paper, shows that the neutron induced fission of americium-241, like its capture product, americium-242(m), has a very high neutron multiplicity, meaning that it is also an excellent breeder fuel under these conditions:

Even limited to the left most region of this graphic, which is actually the region in which "fast" reactors work, the multiplicity is shown to be above 3 neutrons/fission. The graph indicates that were the fusion people ever able to run a fusion reactor, where neutrons emerge with an incredible 14 MeV energy, and chose to use a hybrid approach fusion/fission approach to solve the unaddressed problem of heat transfer, americium could provide very high neutron fluxes to do incredible amounts of the valuable work that neutrons can do.
The next question is "How much Americium is Available?"
Kessler - who writes frequently on the subject of denaturing nuclear materials to make them useless for weapons applications - addressed this issue about 12 years ago in this paper: G. Kessler (2008) Proliferation Resistance of Americium Originating from Spent Irradiated Reactor Fuel of Pressurized Water Reactors, Fast Reactors, and Accelerator-Driven Systems with Different Fuel Cycle Options, Nuclear Science and Engineering, 159:1, 56-82.
According to the IAEA calculations he referenced in this paper, as of 2005, there were about 106 MT in used nuclear fuel all around the world. During storage of used nuclear fuel, especially MOX fuel, but not limited in any sense to it, the amount of americium rises because of the decay of its precursor, plutonium-241, Pu-241, which has a half-life of 14.29 years, and which invariably accumulates as a nuclear reactor operates. Ten years after removal from an active core, about 61.6% of the Pu-241 in the fuel will still be present, with the balance having decayed to Am-241. Because of anti-nuclear fear and ignorance, which is quite literally killing the world we know and love, we now have unprocessed used nuclear fuel that has been stored for 40 years or more, without having been processed, albeit this without killing anyone. Forty year old used nuclear fuel will still contain 14.4% of the Pu-241 originally in it, but will obviously have more americium than it did when it was removed from the reactor. (The half-life of Am-241 is 432.6 years.) It seems reasonable to me that we may have more than 200 MT of americium available. This is not a lot of material, but it is significant. The energy content of this much americium, fully fissioned, is very roughly equal to about 10% to 20% of the annual energy demand of the United States. This much americium is certainly not enough to address any more than a tiny fraction of the completely unaddressed climate crisis, but on the other hand, the high neutron multiplicity can certainly accelerate the rate at which we can accumulate fissionable nuclei that are the only viable option for doing anything about climate change. (And let's be clear. Right now we are doing less than nothing to address climate change.) The amount of americium available to use is certainly enough to run 10 to 20 nuclear reactors running exclusively on americium.
Kessler's paper offered some calculations of the isotopic vectors of the Americium available in different fuel scenarios and presented them graphically. In connection with the thermal neutron spectrum graphics he writes:
...In options C, D, and E, either reenriched reprocessed uranium ~RRU! ~option C! or natural uranium or RRU both together with plutonium options D and E! are used…
...In options F and G, RRU or thorium is mixed with plutonium and MAs in order to incinerate both plutonium and MAs in PWRs.
Figure 2:

The graphic refers to a 10 year cooling period, and again, many used nuclear fuels are much older, with the result that the percentage of Am-241 in these cases would be more enriched in Am-241 with respect to the other isotopes.
Kessler offers similar analysis in various fast neutron cases which I will not discuss here. Very few of these types of reactors have operated commercially, although the advent of small modular "breed and burn" reactors, which Kessler does not address, will likely change this. Kessler also doesn't address a case which in my imagination would be superior to all solid fuel cases, specifically liquid metal fuels, a case that was abandoned in the 1960s because of the relatively primitive state of materials science in those days but is certainly worth reexamining 60 years later, given significant advances in that science.
Anyway, in writing this post, and researching it, I have convinced myself once in for all, that my previous feeling that it was a disgrace to let Pu-241 decay into Am-241 was wrong. I have changed my mind: Am-241 is more valuable than Pu-241, even accepting the high value of Pu-241.
This brings me to the Science paper referenced at the outset of this post:
Americium in its chemistry is dominated by the +3 oxidation state, and in this state, it behaves very much like the lanthanide f elements. Significant quantities of the lower lanthanide f elements are formed via nuclear fission, from lanthanum itself up to and including gadolinium.
The process described in the above referenced Science paper uses electrochemistry to make americium behave more like uranium, neptunium, and plutonium, in exhibiting oxidation states higher than +3, and thus having far different properties than the lanthanides.
From the paper's introduction:
Partitioning of Am from the lanthanides is arguably the most difficult separation in radiochemistry. The stable oxidation state of Am in aqueous, acidic solutions is Am(III). With its ionic radius comparable to the radii of the trivalent lanthanide ions, its coordination chemistry is similar, leaving few options for separation. One approach is the use of soft donor ligands that exploit the slightly more diffuse nature of the actinide 5f-orbitals over the harder lanthanide 4f-orbitals. This provides a stronger, more covalent bond between actinides and N-donor ligands. Notable progress has been made in complexation-based strategies, but considerable challenges have been encountered when attempting to adapt their narrow pH range requirements for process scale-up, stimulating efforts to find alternatives. Another approach is oxidation and separation using the higher oxidation states of Am (5, 6). Unlike the lanthanides, with the exception of the ceric cation, Am(III) can be oxidized and forms [AmVO2]+ and [AmVIO2]2+ complex ions in acidic media. The high oxidation state ions could be separated from the lanthanides by virtue of their distinct charge densities using well-developed solvent extraction methods (4)...
Although solvent extraction method are in fact, well developed, it does not follow that they are the best approach to separations of the elements in used nuclear fuel. If I were to look at electrochemical oxidation of americium, I would also look at electrochemical based separation, including an analogue to the separation of nucleic acids and proteins, electrophoretic migration through gels, or liquid/liquid, gel, or solid based ion selective membranes.
The beauty of electrochemistry is that it avoids the use of reagents. the wide use of primitive solvent extraction methods in the mid to late 20th century led to problematic situations like the Hanford tanks about which very stupid anti-nukes carry on endlessly even as they completely ignore, with contempt for humanity, the millions upon millions of people killed every damned year, at a rate if about 19,000 human beings per day, from dangerous fossil fuel waste.
More excerpts of text in the paper:
...Oxidizing Am(III) in noncomplexing media is hampered by the high potential for the intermediate Am(IV/III) couple (Fig. 1) (15). Only a limited number of chemical oxidants, including persulfate and bismuthate, have been explored for this purpose (16). Oxidation by persulfate gives sulfate as a by-product, which complicates subsequent vitrification of the waste (17). Bismuthate suffers from very low solubility, necessitating a filtration step that complicates its removal (16). Adnet and co-workers have patented a method for the electrochemical generation of high-oxidation-state Am in nitric acid solutions (18), based on earlier results by Milyukova et al. (19, 20), who demonstrated Am(III) oxidation to Am(VI) in acidic persulfate solutions with Ag(I) added as an electron transfer mediator with Eo[Ag(II/I)] = 1.98 V versus SCE...
The fact that the authors are embracing the cultural mentality of so called "nuclear waste," rather than the recovery of valuable nuclear materials, has no bearing on the quality of their science. I just have to say that.
The redox potentials of americium:

The caption:
The authors proposed the use of a new type of electrode, an organic/indium tin oxide electrode, to oxidize americium. Here is a a cartoon representation of this electrode:

The caption:
(Left) A simple molecular illustration. (Right) A density functional theory–optimized p-tpy structure with pyridine rings oriented to show the potential tridentate bonding motif, ideally placed on a surface.
A word on indium tin oxide: This compound is found in pretty much every touch screen in the world. It is also widely used in some types of solar cells. In the past, at DU, I used to engage a dumb "renewables will save us" anti-nuke in discussion of the fact - facts matter - that the world supply of indium is limited, and therefore any so called "renewable energy" technology based on it is, um, well, not in fact "renewable." Ultimately this person bored me to death, and I put him or her on my lovely "ignore" list, since there is no value in engaging people who simply repeat distortions and outright lies over and over, in a Trumpian fashion. The fact remains supplies of indium are limited. It is considered a "critical material" of concern to people who study such things. The world does risk running out of it. The difference, of course, between cell phones and solar cells and nuclear fuel reprocessing plants do not require large amounts of mass, since the environmental superiority of nuclear fuel to all other energy forms derives entirely from its enormous energy to mass ratio. In fact, indium is a minor fission product found in used nuclear fuel, and more than enough can easily be recovered from such a fuel to make these electrodes. I am not endorsing these particular types of electrodes by the way; I'm not sure that nitric acid solutions are in fact the best approach to nuclear fuel reprocessing; there are strong arguments that they are not. The point is, that in comparison to the requirements of solar cells and cell phones, the requirement for indium in this case, were it to go commercial - and it never may do so - is trivial.
Some graphics on the electrochemical results obtained in working with the electrode:

The caption:
(Left) Am speciation measured by visible spectroscopy in a 1-cm cuvette at an applied potential of 1.8 V versus SCE. The appearance of Am(V), concurrent loss of Am(III), and overall mass balance are plotted. (Right) Electrochemical Am oxidation scheme involving a p-tpy–derivatized electrode.
The authors remark on the effect of radiation on these solutions:
Radiolysis of water by Am generates one-electron reducing agents such as H atoms and two-electron reducing agents such as hydrogen peroxide, as well as other redox transients (35). The concentration of radiolysis products varies linearly with total Am concentration, with zero-order reduction kinetics observed for the appearance or disappearance of Am species. Under these conditions, rate constants for these Am species during autoreduction can therefore be derived from the slopes of concentration-time plots (36, 37).
Radiolytically produced one-electron and two-electron reductants provide independent pathways for Am(VI) reduction, with an overall rate constant for Am(VI) loss of 23.4 × 10?6 s?1 (fig. S10). The Am(IV) produced from the two-electron reduction of Am(VI) by radiolytic intermediate or intermediates, presumably H2O2, rapidly disproportionates to Am(V) and Am(III). The reduction of Am(V) to Am(IV) or Am(III) is slow on this time scale (fig. S10).
It seems to me, perhaps naively, that this issue might be addressed by continuous separation, by methods to which I alluded above, to continuous separations, driving the equilibrium. Part of the problem might be addressed by putting beta emitting species - which in real life would be present anyway - in the solution, but no matter.
Some more commentary:
Figure 4:

The caption:
(Left) Am speciation as measured by visible spectroscopy in a 50-cm waveguide. (Right) Visible spectra of species before controlled potential electrolysis and after 13 hours of electrolysis with highlighted speciation changes.
The authors conclude:
This is an interesting little paper. I very much enjoyed going through it as well as thinking a little more deeply about the entire subject of americium, which might prove a valuable tool in saving the world via the use of nuclear energy.
While considering all of this, I have to say what's always on my mind:
To my way of thinking, opposition to nuclear energy - even though such opposition appears often on my end of the political spectrum - is criminally insane.
Right now, hurricanes are marching, year after year, all over the United States and other parts of the world. Vast forests, farms and homes have burned all over the Western United States this year, droughts are destroying crops as are things like derechos. We do not record the number of people killed by extreme heat events, but the scientific literature suggests these numbers are rather large, and we have seen the most extreme temperatures ever recorded in 2020. Insects that spread infections are appearing at ever higher latitudes...
The list of the scale of what we are experiencing from climate change goes on and on and on...
And yet...and yet...and yet...I hear from people that "nuclear power is too dangerous." Compared to what?
Let's be clear on something, OK? Opposition to nuclear energy is as ridiculous and as absurd as refusing to wear masks in the Covid-19 epidemic because of political orthodoxy; similarly it kills people. Many of us on the left should examine and confront our own anti-science political orthodoxy, much as old white men like myself need to examine and confront our suppressed racist rationalizations.
Questioning ourselves is something we all must do; because we cannot hope to struggle to achieve the status of being moral human beings without doing so.
Nuclear energy need not be entirely risk free to be vastly superior to everything else; it only needs to be vastly superior to everything else, which it is. It is immoral to oppose nuclear energy.
I trust that you will have a safe, yet rewarding weekend.
How Trump damaged science -- and why it could take decades to recover.
This is a news item in the current issue of the scientific journal Nature: How Trump damaged science — and why it could take decades to recover. (Jeff Toleffson, Nature News October, 6, 2020)
I don't believe a subscription is required.

Some excerpts from this rather long news story, necessarily long since so much damage has been done.
Some excerpts:
US President Donald Trump’s rally in Henderson, Nevada, on 13 September contravened state health rules, which limit public gatherings to 50 people and require proper social distancing. Trump knew it, and later flaunted the fact that the state authorities failed to stop him. Since the beginning of the pandemic, the president has behaved the same way and refused to follow basic health guidelines at the White House, which is now at the centre of an ongoing outbreak. As of 5 October, the president was in a hospital and was receiving experimental treatments.
Trump’s actions — and those of his staff and supporters — should come as no surprise. Over the past eight months, the president of the United States has lied about the dangers posed by the coronavirus and undermined efforts to contain it; he even admitted in an interview to purposefully misrepresenting the viral threat early in the pandemic. Trump has belittled masks and social-distancing requirements while encouraging people to protest against lockdown rules aimed at stopping disease transmission. His administration has undermined, suppressed and censored government scientists working to study the virus and reduce its harm. And his appointees have made political tools out of the US Centers for Disease Control and Prevention (CDC) and the Food and Drug Administration (FDA), ordering the agencies to put out inaccurate information, issue ill-advised health guidance, and tout unproven and potentially harmful treatments for COVID-19.
“This is not just ineptitude, it’s sabotage,” says Jeffrey Shaman, an epidemiologist at Columbia University in New York City, who has modelled the evolution of the pandemic and how earlier interventions might have saved lives in the United States. “He has sabotaged efforts to keep people safe.”
The statistics are stark. The United States, an international powerhouse with vast scientific and economic resources, has experienced more than 7 million COVID-19 cases, and its death toll has passed 200,000 — more than any other nation and more than one-fifth of the global total, even though the United States accounts for just 4% of world population...
...As he seeks re-election on 3 November, Trump’s actions in the face of COVID-19 are just one example of the damage he has inflicted on science and its institutions over the past four years, with repercussions for lives and livelihoods. The president and his appointees have also back-pedalled on efforts to curb greenhouse-gas emissions, weakened rules limiting pollution and diminished the role of science at the US Environmental Protection Agency (EPA). Across many agencies, his administration has undermined scientific integrity by suppressing or distorting evidence to support political decisions, say policy experts.
“I’ve never seen such an orchestrated war on the environment or science,” says Christine Todd Whitman, who headed the EPA under former Republican president George W. Bush.
This is from Christie Todd Whitman, who in the Bush administration was certainly a previous "worst ever" administrator of that agency, refusing to do a damned thing about climate change. Even a Republican with as a pathetic view of environmental issues can be outraged by Trump.
“It’s terrifying in a lot of ways,” says Susan Hyde, a political scientist at the University of California, Berkeley, who studies the rise and fall of democracies. “It’s very disturbing to have the basic functioning of government under assault, especially when some of those functions are critical to our ability to survive.”...
There's plenty here, read it and weep...
Why Nature needs to cover politics now more than ever
An Editorial in Nature has recently discussed why scientists must be politically engaged: Why Nature needs to cover politics now more than ever.
It's open sourced; no subscription is required.
For convenience, some excerpts:
This week, Nature reporters outline what the impact on science might be if Joe Biden wins the US presidential election on 3 November, and chronicle President Donald Trump’s troubled legacy for science. We plan to increase politics coverage from around the world, and to publish more primary research in political science and related fields.
Science and politics have always depended on each other. The decisions and actions of politicians affect research funding and research-policy priorities. At the same time, science and research inform and shape a spectrum of public policies, from environmental protection to data ethics. The actions of politicians affect the higher-education environment, too. They can ensure that academic freedom is upheld, and commit institutions to work harder to protect equality, diversity and inclusion, and to give more space to voices from previously marginalized communities. However, politicians also have the power to pass laws that do the opposite.
The coronavirus pandemic, which has taken more than one million lives so far, has propelled the science–politics relationship into the public arena as never before, and highlighted some serious problems. COVID-related research is being produced at a rate unprecedented for an infectious disease, and there is, rightly, intense worldwide interest in how political leaders are using science to guide their decisions — and how some are misunderstanding, misusing or suppressing it...
...Perhaps even more troubling are signs that politicians are pushing back against the principle of protecting scholarly autonomy, or academic freedom. This principle, which has existed for centuries — including in previous civilizations — sits at the heart of modern science.
Today, this principle is taken to mean that researchers who access public funding for their work can expect no — or very limited — interference from politicians in the conduct of their science, or in the eventual conclusions at which they arrive. And that, when politicians and officials seek advice or information from researchers, it is on the understanding that they do not get to dictate the answers...
... Last year, Brazil’s President Jair Bolsonaro sacked the head of the country’s National Institute for Space Research because the president refused to accept the agency’s reports that deforestation in the Amazon has accelerated during his tenure. In the same year, more than 100 economists wrote to India’s prime minister, Narendra Modi, urging an end to political influence over official statistics — especially economic data — in the country.
And just last week, in Japan, incoming Prime Minister Yoshihide Suga rejected the nomination of six academics, who have previously been critical of government science policy, to the Science Council of Japan. This is an independent organization meant to represent the voice of Japanese scientists. It is the first time that this has happened since prime ministers started approving nominations in 2004...
An Enantioefficient Synthesis of Ergocryptine w/Direct Synthesis of the (+) Lysergic Acid Component.
The paper I'll discuss in this post is 16 years old. It is this one: Enantioefficient Synthesis of ?-Ergocryptine:? First Direct Synthesis of (+)-Lysergic Acid (István Moldvai, Eszter Temesvári-Major, Mária Incze, Éva Szentirmay, Eszter Gács-Baitz, and Csaba Szántay The Journal of Organic Chemistry 2004 69 (18), 5993-6000)
This is the structure of ergocryptine as drawn in this paper:

When this molecule is brominated, using N-bromosuccinimde, it is called bromocryptine, which is a drug used to treat Parkinson's disease, a few types of substance abuse diseases, and, interestingly, Type 2 diabetes.
Bromocryptine:

Recently, in this space, I referred to the latter molecule in a paper about the relationship between microbial fuel cells and Parkinson's disease in connection with electron transfer, and I remarked that I'd informed my sons and my son's girlfriend, that the structure of the upper ring system, which is obviously peptide derived from the amino acids valine, leucine and proline, had a hemiacetal structure.
That post (uncorrected) is here: Electron Shuttling in Parkinson's Disease Elucidated by Microbial Fuel Cells.
Thankfully I was corrected by a correspondent, our leader in this forum in fact, who noted that this is in fact not a hemiacetal, but is rather an orthohemiaminal.
To be frank, I can't recall running across too many orthoaminals, but I assumed, correctly, that somewhere that there was a synthesis of this molecule, which is a derivative of lysergic acid, the famous precursor to LSD which was discovered at Sandoz by Albert Hoffman, whose serious scientific work work on ergot alkaloids includes a synthesis of the upper ring system, a paper which I will cite below. The paper cited above at the outset includes a direct synthesis of lysergic acid and an improved synthesis of Hoffman's synthesis of the upper ring system.
Orthoaminals seem to be relatively rare in the literature, a quick search in Google scholar in my hands produced less than 100 hits. It is thus somewhat surprising that Hoffman's generation of that structure was not all that elaborate from what I can pick out of the German text.
First the introductory text from the paper cited at the outset:
The first synthesis of racemic lysergic acid was effected by Woodward and Kornfeld in 1956.3,16 One of their main problems was to prepare ring C from the otherwise easily accessible 3-indolepropionic acid (3), since the ring closure of the corresponding acid chloride occurred at the more reactive pyrrole ring instead of the benzene ring. Thus the Woodward group reduced the pyrrole ring, the amine was protected by benzoylation, and thereafter the ring closure took place regioselevtively as desired. The drawback of this approach is that sooner or later the pyrroline moiety must be reoxidized to a pyrrole ring. It is difficult to perform an enantioefficient synthesis as well, since the method involves introduction of an unnecessary chiral center by the reduction. The earlier described resolution of racemic compound needs further and rather inconvenient steps.4 So far the total synthesis of (±)-2a has been achieved by nine groups, but the number of publications dealing with the synthetic efforts is much higher. Among these approaches one can find about a dozen methods trying to construct the ergoline ring, some of which were successful; others remained at the level of attempt.5 Seven of the nine successful syntheses used the reduced indoline derivative as the starting compound. Oppolzer et al.3 performed the first total synthesis avoiding the reduction step, but their procedure again cannot be scaled up. A second approach to the racemic acid was published recently.6
We decided to construct the ergoline skeleton starting from indole, thus avoiding the reoxidation problem, and at the same time making an enantioefficient synthesis possible.7
An ideal starting material was the so-called Uhle's ketone (4a) having the intact indole ring, although the original synthesis of 4a is rather tedious. Uhle commenced with acetylation and subsequent bromination, and he claimed that the derived bromo-derivative 4b could be subjected successfully to a substitution reaction with various amines.9 Early in the seventies Bowman and co-workers reinvestigated a few results of Uhle's synthesis and established that one of the key steps, alkylation of several types of amines with 4b, always failed.10 The approach starting from 4a by Stoll used the Stobbe condensation as a key step, but the reaction sequence could not be carried out,11 thus the synthesis of lysergic acid starting from Uhle's ketone remained a challenge.
How they did it:

The caption:
a?Reagents and conditions:? (a) (1) powdered KOH + Piv-Cl, CH2Cl2 + THF, (2) SOCl2, (3) AlCl3 + ClCH2COCl, CH2Cl2 (43%, overall); (b) ref 15 (85%); (c) HO(CH2)2OH, p-TSA, benzene, reflux, 6 h (81%); (d) MeNH2, CHCl3, 10?15 °C, 3?4 h (88%); (e) aq HCl (1 M), acetone, rt, 3 h (97%).

The caption:
a?Reagents and conditions:? (a) 5 + 4d, toluene, 48 h, rt (35%); (b) 5 + 4g, THF, 24 h, rt (56%); (c) MeNH2, benzene, 10?15 °C, 1 h (80%); (d) aq HCl (6 M), 35?40 °C, 1 h then (e) 6c in CHCl3, LiBr + TEA, 0?5 °C, 12 h (60%, 2 steps); (f) ( ? )-dibenzoyl-l-tartaric acid, CH3CN + H2O (1:1) (38%).

The caption:
a?Reagents and conditions:? (a) (+)-7 + 8, t-BuOK, THF + t-BuOH, 0 °C, 20 min then + H2O, ?5 °C (77%); (b) aq HCl (2 M), reflux, 30 min (13%); (c) NaOMe, MeOH, 70?75 °C, 30 min (70%); (d) HCl/MeOH (6.7 M), 75?80 °C, 45 min (72%); (e) aq NaOH (5 M), MeOH, 70?80 °C, 2.5 h then aq HCl (6 M) to pH 6.5 (54%).
When I was a kid, I used to do a fair amount of amino acid related synthetic chemistry, and so I tend to see amino acids in everything. I always saw the tryptophan in the structure of lysergic acid, and I remember chatting with a friend of mine the way one does in the lab and speculating that they cyclization to what I now know as Uhle's ketone could be accomplished by having the tryptophan side chain acylate the benzogroup of the indole ring. This would retain the necessary stereochemistry at the nitrogen, free of charge. I was a stupid and naïve kid, since there is no way to assure the regiochemistry of actually accomplishing this, but it's not like it was even a remotely serious conversation.
It turns out however, as is referenced in this paper, that Rappaport group did synthesize a stereochemically pure Uhle's ketone using a serine starting material: Enantiospecific Synthesis of (R)-4-Amino-5-oxo-1,3,4,5-tetrahydrobenz[cdindole, an Advanced Intermediate Containing the Tricyclic Core of the Ergots] (Clarence R. Hurt, Ronghui Lin, and Henry Rapoport The Journal of Organic Chemistry 1999 64 (1), 225-233)
Here is the route, beginning with the (complex) synthesis of bromotryptophan in such a way as to assure regiospecificity for the cyclization.

The caption:
The serine in this scheme is the molecule numbered 12; it is protected as a MOM (methoxymethyl) acetal of formaldehyde. (I should have used that protecting group more often; but I was a stupid kid).
Uhle's ketone:

The caption:
a?Both (±) and (+) compounds were prepared; only the (+) form is shown.
Anyway, about that orthohemiaminal. I kind of thought it would be very difficult to make, but it looks like it sort of falls out in this elegant synthesis by Albert Hofmann and his coworkers. The text is in German:
Die Synthese der Alkaloide der Ergotoxin‐Gruppe. 70. Mitteilung über Mutterkornalkaloide (P. A. Stadler St. Guttmann H. Hauth R. L. Huguenin Ed. Sandrin G. Wersin H. Willems A. Hofmann, Helv Chim Acta Volume 52, Issue 6 1969 Pages 1549-1564)

In this picture, the awkwardly drawn molecule 10 with the ridiculously long amide bond spontaneously arranges to the orthohemiaminal, 11.
The text describing this reaction is in German, so one can either look at the pictures or muddle through translating the German.
The German Text:
My translation:
The emphasis is mine.
So much for the difficulty of forming a orthohemiaminal.
On a day like this, where one thinks, given the nutcase in the White House, that one must be hallucinating, must be on a bad trip, it was probably appropriate to stumble upon some lysergic acid chemistry while trying to think about electron transfer in Parkinson's disease and in microbial fuel cells.
Have a nice day tomorrow.
Biden is going to need to seriously disinfect the White House when he moves in.
He should also take care of any Covid residues.
Using Corn Cob Derived Biomaterials to Remove Heavy Metals and Kill Bacteria in Contaminated Water.
The paper I'll discuss in this post is this one: Polyethylenimine-Grafted-Corncob as a Multifunctional Biomaterial for Removing Heavy Metal Ions and Killing Bacteria from Water (Qin Meng, Shengdong Wu, and Chong Shen Industrial & Engineering Chemistry Research 2020 59 (39), 17476-17482).
With a little bit of googling, it appears to me that the world produces about 1.1 billion tons of corn each year, presumably measured as grain. According to the introduction of the above cited paper, which I will excerpt, corn cobs, which are generally discarded, account for a mass 1/4 as large as the grain itself, somewhere around 250 million tons. From a table in the paper, that I'll reproduce from the paper, it appears that corncobs are about 43% carbon, meaning that somewhere around 100 to 110 million tons of carbon can be obtained from corncobs.
While we are engaged in Godotian waiting for the grand so called "renewable energy" nirvana that has not come, is not here, and won't come, we are currently dumping about 35 billion tons of the dangerous fossil fuel waste carbon dioxide into the planetary atmosphere.
The interesting product discussed in this paper will not sequester all that much carbon in use, even if - as surely will not happen - all the corn cobs in the world were utilized to make the biomaterial discussed herein. Nevertheless this is carbon captured from the air, and every little bit counts. In theory, at least, carbon captured from corn cobs and sequestered as value added products might remove a billion tons every decade, not all that much, but nothing at which to sneeze. Thus this is an interesting little paper.
The corn cobs in this paper are functionalized, as noted in the title, with polyethylenimine, which is a polymer synthesized by chain ring opening of aziridine, the simplest nitrogen organic heterocycle, a cyclic molecule having two carbons, five hydrogens and one nitrogen. Aziridine is generally made via various routes from ethylene. All, or almost all, of the world's ethylene is obtained from dangerous fossil fuels. However many other starting materials are certainly possible on an industrial scale. Syngas is a mixture of hydrogen and carbon oxides - generally the monoxide - which can be made from pretty much any carbon containing material and water, including carbon dioxide with the agency of heat, nuclear heat if one is seeking a carbon free route. There are many routes to ethylene from syngas. My personal favorite proceeds via the intermediate dimethyl ether, which I personally believe should be the core chemical portable energy medium in a sustainable world. It is described here: Direct Conversion of Syngas into Methyl Acetate, Ethanol, and Ethylene by Relay Catalysis via the Intermediate Dimethyl Ether (Zhou et al., Angew. Chem. Int. Ed. 2018, 57, 12012 –12016)
Ethylene can also be made via the partial hydrogenation of acetylene, and acetylene can be made from multiple carbon sources by many means, notably heating a carbon source with calcium metal, albeit with varying degrees of efficiency. The calcium hydroxides produced in such a process can be useful for carbon dioxide capture from either air or from combustion gases or reformer gases.
Thus it is theoretically possible to derive the corn cob derived product described here entirely from carbon dioxide in the air, although any polyethylenimine used for these purposes would merely sequester dangerous fossil fuel derived carbon.
Here is the cartoon accompanying the abstract of the paper:

From the paper's introduction:
By now, corncob-based biochars, corncob mixtures, and even corncob itself have been developed into biosorbents for removal of heavy metal ions from aqueous solution. For example, calcined corncob biochars showed maximum adsorption capabilities of less than 40 mg/g for Cu2+,(7) Pb2+,(8) and Cd2+,(8) while the acrylonitrile modification on the biochars improved the Cd2+ adsorption capacity to 85.6 mg/g.(2) In addition, a mixture of tea waste, corncob, and sawdust had adsorptive capacities of 39.5, 94.0, and 41.5 mg/g for Cu2+, Pb2+, and Cd2+.(9) Interestingly, Garg et al. reported that corncob itself could remove 105.6 mg/g of Cd2+ from aqueous solution,(10) even more potent than the biochars and mixtures. Nevertheless, the adsorptive capabilities of these biosorbents are not high because the mechanism was dominated by physisorption via the weak van der Waals force between the metal ions and corncob surface(8) due to the lack of functional groups on the sorbents.
Polyethylenimine (PEI), a polymer that contains a large amount of primary and secondary amine side groups, can bind to heavy metal ions via chelating forces.(11) The strong binding forces of chelation attributed to the high adsorptive capability (>100 mg/g) of PEI modified materials such as bacterial cellulose,(12) P84 nanofiltration membrane,(13) chitin,(14) and magnetic nanoparticles(15) to heavy metal ions. In addition, PEI also displayed wide-spectrum antimicrobial effects(16) due to its cation property, so that the PEI-grafted materials (e.g., polyurethane ureteral stents) showed potent antibacterial ability in medical applications.(17,18) Economically, PEI is a bulk chemical that is much cheaper than other materials such as silver nanoparticles.
Because of these properties of PEI, this paper aims to develop a multifunctional material via grafting PEI on corncob. The PEI-modified corncob is expected to perform functions like removing heavy metal ions and killing bacteria. As the two aspects are critical and usually coexist in treatment of polluted water, multifunctional PEI-g-OC should be a useful biomaterial in the application of water treatment, e.g., portable devices for water purification. The PEI-g-OC performs significant advantages on cheapness, lightweight and environmental-friendliness over chemical synthetic materials.
A cartoon about the route to the product, a photograph of the starting materials, intermediate and product, and some spectra:

The caption:
A table of the elemental composition of the final product and the intermediates, giving a feel, as mentioned above, for the carbon sequestration capability of these materials:

Copper, for which toxicity is concentration dependent, is pretty much found in all potable water from piping, cadmium and lead to varying degrees from historic solders and or leachate from landfills, cadmium from discarded nickel cadmium batteries, and - increasingly in the future - from discarded CIGS type photovoltaic devices, both portable (as in old calculators and toys) and/or solar cells.
An idea of the capacity of these materials for heavy metals, in this case cadmium, copper and lead:

The caption:
Langmuir and Freundlich isotherms, which are technical measurements of evaluating surface chemistry:

The caption:
The antibacterial properties of the PEI impregnated corn cob material was determined against E. coli, P. aeruginosa, and S. aureus:

The caption:

The caption:
The authors determined that the product was reusable:

The caption:
The paper's conclusion:
It is notable that products like these that capture metals can be utilized to create low grade ores. The inexhaustibility of uranium resources is tied to precisely such an approach.
It's an interesting little paper.
I wish you a safe, healthy and enjoyable Sunday afternoon.
Electron Shuttling in Parkinson's Disease Elucidated by Microbial Fuel Cells.
The paper I'll discuss in this post is this one: Deciphering Electron-Shuttling Characteristics of Parkinson’s Disease Medicines via Bioenergy Extraction in Microbial Fuel Cells (Chen et al, Industrial & Engineering Chemistry Research 2020 59 (39), 17124-17136.
Since my regular journal reading list - the reading list for pleasure, as opposed for my paid work - often includes journals focusing on issues relating to energy and the environment, I often come across papers relating to microbial fuel cells. I tend not to read these papers in any level of detail, since my main focus is on carbon dioxide issues and high temperature thermal schemes, but my general feeling is that they may prove to be a way to recover some energy in water treatment systems while simultaneously cleaning up the water.
The current issue of Industrial & Engineering Chemistry Research is about biomass utilization, a topic which is of very high interest to me, since it is one route - certainly not the only route - to capture of carbon dioxide from the air, a topic which will be of extreme importance in the future given our ongoing failure to do anything even remotely practical to address climate change.
In spite of my low passing interest in microbial fuel cells, this particular article caught my eye because my oldest son, a designer/artist, has an independent interest in neurobiology as it relates to perception and in general, to mood and to consciousness itself. In addition my youngest son's girlfriend is studying neurobiological psychology. Also any paper that intrinsically brings together two very disparate scientific issues is immediately interesting.
From the introductory text:
... Moreover, for dihydroxyl (diOH)-bearing aromatics, compared to meta-isomers, ortho- or para-dihydroxyl (diOH) substituent-bearing phenolics possessed more promising electrochemical activities for electron-shuttling.(5,6) For example, the literature(7) further mentioned that accumulation of azo dye-decolorized metabolites (DMs) could catalytically stimulate the efficiency of electron transfer, thereby enhancing the bioelectricity generation of MFCs. However, microbial decolorization of azo dyes would generate aromatic amine (?NH2) intermediates,(8) inevitably leading to possible concerns of secondary pollution to the environment...
...Recently, the study(10) has also evaluated ortho-diOH-substituents (e.g., o-diOH-bearing dopamine (DA) and epinephrine (EP)), showing that such neurotransmitter-related chemicals could significantly mediate electrochemical properties, thereby effectively promoting bioelectricity generation in MFCs. It was also suspected that such electron-mediating capabilities were strongly associated with neurotransmission among neurons, muscle cells, or gland cells. For example, DA is an endogenous hormone and neurotransmitter in the human brain and body. It is released from presynaptic neurons to synaptic cleft and binds to postsynaptic receptors to causes actions of postsynaptic cells, in turn promoting transmission among postsynaptic cells.(11)...
...The major families of drugs to treat PD are dopamine agonists, Levodopa, and monoamine oxidase inhibitors (MAO-B inhibitors).(16,17) They are applied to different nerve conduction or neurotransmitting pathways to reduce symptoms of PD. In fact, as Levodopa and dopamine are electron-shuttling chemicals, it was thus proposed that the involvement or initiation of DA-associated chemicals with high bioelectrochemical-catalyzing activities seemed to be significant for disease medication. That was why this study selected seven representative medicines used in clinical treatment for comparative assessment (Figure 1; Table S1)...
Figure 1:

The caption:
I sent my sons and my youngest son's girlfriend the following commentary when I emailed this paper to them:
The three amino acids in this interesting ring system are valine and isoleucine and proline. The carboxylic acid in proline has been reduced to an aldehyde and then formed into a structure called a "hemiacetal, connected to an oxygen from an oxidized form of valine, alpha hydroxyvaline, making the valine a "aminal." These types of lysergic acid/peptoid structures are very common in the ergot alkaloids, from which several major neuroactive, including neuromuscular active, drugs have been developed, including ergotamine, and methasergide for chronic headaches, ergonovine and methergine to induce labor, and of course, the chemically brominated bromocriptine, utilized in parkinson's.
LSD, and a few other hallucinogenic molecules are also derivatives of lysergic acid, of course.
The point is that all of these molecules act on neurotransmitters.
The microbial fuel cells utilized an electroactive organism, Aeromonas hydrophilia, which was originally obtained from a river in Taiwan. It apparently has been investigated for it's ability to decolorize waste water from dyeing plants in the textile industry.
Some more text this from the main body of the paper:
The authors found that L-Dopa, dopamine, and APO all gave enhanced electrochemical signals in the microbial fuel cell.
Some more figures from the text:

The caption:

The caption:
The authors utilized a high resolution mass spec, a Thermo Q Exactove Plus - an orbitrap mass spec - to study the products of the APO, apomorphine, in the fuel cell.

The caption:
The authors studied the power density of the microbiological fuel cells in the presence of Parkinson's medications. Note that the units give a feel for the required size for these types of devices:

The caption:
The authors studied various hydroxybenzenes, a few of which are available from lignins, suggesting that the "black liquor" of paper making, which contains lignins, may be utilized in microbial fuel cell type systems to clean up the black liquor if the base can be neutralized. It should be said that lignins, I believe, will have many future uses in a post dangerous fossil fuel world, should one ever come to exist. I should note in the context of this paper, that one of the trihydroxybenzenes listed here, gallic acid, which can be obtained from wood, is a potential synthetic precursor to the neurologically active hallucinogen mescaline, in six - possibly fewer - steps, the point being that hydroxylated benzenes screw with your nerves. (The street drug known as Ecstasy or "Molly" is also a hydroxybenzene derivative, as is vanilla.)

The caption:

The caption:
A brief excerpt of the conclusion:
An interesting and different approach to drug screening I think. Whether it would prove superior to other screening tools, I can't say. I haven't worked in neuroactive drug development, except for some very remotely related work on Alzheimer's medications and a few purely analytical programs for drugs utilized in PD.
Have a pleasant Sunday.
My spell checker is always acting smarter than I am, so I beat the crap out of it.
I typed "highly sulfonated quinolindinium/benzyloxazolidinium copolymeric catalyst" in a note to myself and the sucker choked.
I sometimes get tired of its smart assed spelling and grammar corrections. It's like that spelling bee winner in the fifth grade who liked to stick her tongue at you.
It's probably sulking somewhere in its solid state silicon brain.
Rate of mass loss from the Greenland Ice Sheet will exceed Holocene values this century.
The paper I'll discuss in this post is this one: Rate of mass loss from the Greenland Ice Sheet will exceed Holocene values this century (Jason P. Briner, Joshua K. Cuzzone, Jessica A. Badgeley, Nicolás E. Young, Eric J. Steig, Mathieu Morlighem, Nicole-Jeanne Schlegel, Gregory J. Hakim, Joerg M. Schaefer, Jesse V. Johnson, Alia J. Lesnek, Elizabeth K. Thomas, Estelle Allan, Ole Bennike, Allison A. Cluett, Beata Csatho, Anne de Vernal, Jacob Downs, Eric Larour & Sophie Nowicki, Nature volume 586, pages 70–74 (2020))
So called "renewable energy" hasn't saved the world; it isn't saving the world; it won't save the world. I have nothing more to say about the literally pyrrhic apparent triumph of the antinukes than what it says on the AAAS t-shirt distributed this year says: Facts are facts.
Since this paper which is a modeling paper, suggests, by fitting the model to the best historical data on the Greenland Ice Sheet what the future of the ice sheet will be, given that we have deliberately chosen not to do anything effective about climate change:
The abstract of the paper is available at the link.
For convenience, an excerpt:
They suggest the data to which they fit their model represents a loss of ice amounting to around 6,000 billion tons of ice per century, 6 trillion tons during the Holocene, which is the current post glacial era in which civilization arose. Using their model, they predict that Greenland will lose between 8,000 billion tons to 35,000 billion tons in the 21st century, greatly exceeding any value recorded in the last 12,000 years.
For those lacking access to the full paper, I'll offer a few excerpts and graphics. From the paper's introduction:
A few excerpts from additional sections:
...Geological observations of GIS change are most abundant during the Holocene14. For this reason, the Holocene has been targeted as a timeframe for simulating GIS history15,16,17,18,19,20. Model simulations so far have been used to assess spatiotemporal patterns of GIS retreat and to constrain its minimum size. Simulated changes in ice volume are largely the product of climatic forcing; palaeo-mass balance is typically modelled using one of the ice-core ?18O time series from central Greenland, which is converted to temperature and precipitation, and scaled across the ice sheet15. Some approaches improve model performance with geological constraints, but climate forcing is still scaled from limited ice-core data, sometimes using prescribed Holocene temperature histories to improve model–data fit16,17. One recent study19 used data averaged from three ice-core sites to adjust palaeotemperatures from a transient climate model, and scaled precipitation from one ice-core accumulation record. All these estimates of mass-loss rates during the Holocene provide important context for projected GIS mass loss, but they have not been extended into the future, making quantitative comparisons uncertain...
We place today’s rates of ice loss into the context of the Holocene and the future using a consistent framework, by simulating rates of GIS mass change from 12,000 years ago to AD 2100. We use the high-resolution Ice Sheet and Sea-level system Model (ISSM), which resolves topography as finely as 2 km (refs. 21,22,23). Our simulations are forced with a palaeoclimate reanalysis product for Greenland temperature and precipitation over the past 20,000 years2. This reanalysis was derived using data assimilation of Arctic ice-core records (oxygen isotopes of ice, and snow accumulation) with a transient climate model (Methods). We account for uncertainty in the temperature and precipitation reconstructions by creating an ensemble of nine individual ISSM simulations that have varying temperature and precipitation forcings2 (Methods). Sensitivity tests using a simplified model in the same domain24 suggest that the range in plausible palaeoclimate forcing, which we use, has a larger influence on simulated rates of ice-mass change than do model parameters such as basal drag, surface-mass-balance parameters and initial state. We compare our simulated GIS extent against mapped and dated changes in the position of the GIS margin3,4...
Some pictures from the text:
Fig. 1: Domain for the ice-sheet model and moraine record of past GIS change in SW Greenland:

The caption:
Fig. 2: Increased and variable GIS mass loss during the Holocene.

The caption:
In figure 3, notice the vertical line on the extreme right of the large graphic. This would be an excellent time to tell me all about how many "Watts" of solar cells are installed in California. Please avoid, since we live in the age of the celebration of the lie, using units of energy, GigaJoules - which matter - in favor of units of peak power - which mean zero at midnight in California.
The annual weekly minimum for carbon dioxide concentrations as measured at Mauna Loa was likely reached last week. The data hasn't been posted, but as I follow these data points weekly, I expect it will come in at about 411.0 ppm +/- 0.2 ppm. In 2010, the annual minimum was reached in the week beginning September 26, 2010. At that time, the concentration of the dangerous fossil fuel waste carbon dioxide in the planetary atmosphere was 386.77 ppm. Read the caption and choose your dot on the vertical line that represents the 21st century, the age of "renewable energy will save us" aka, in my mind, the age of the lie.
Fig. 3: Exceptional rates of ice-mass loss in the twenty-first century, relative to the Holocene.

The caption:
Fig. 4: Substantial change in surface elevation of the GIS over the twenty-first century.

The caption:
Some additional text from the paper:
As of 2018, the humanity was consuming 599.34 exajoules of primary energy per year. 81% of that energy came from dangerous fossil fuels, as opposed to 80% of 420.19 exajoules that were being consumed in the year 2000. Things are getting worse, not better.
Every year, quantities of carbon dioxide added to the atmosphere as dangerous fosssil fuel waste amounts to more than 35 billion metric tons. Land use changes, including those involved in providing so called "renewable energy" - for example the destruction of the Pantanal for ethanol farms - add about another ten billion tons.
To provide about 600 exajoules of primary energy each year, would require, assuming 190 MeV/fission, ignoring neutrinos, completely fissioning about 7.5 thousand tons of plutonium each year. The density of plutonium, in at least one allotrope, is about 19.9 g/ml, depending on the isotopic vector. The size of a cube containing 7.5 thousand tons of plutonium - which could never be assembled as such owing to criticality constraints - is less than 8 meters on a side.
I am often informed by people that "nobody knows what to do with (so called) 'nuclear waste.'" In saying this, it is very clear that these people have never in their wildest imagination ever considered what to do with hundreds of billions of tons of dangerous fossil fuel waste, which is choking the planet literally to death. Of course, their considerations are weak, because the best evidence is that these people can't be bothered to open a scientific paper or a science book on the subject of any kind of waste. Somehow people expect me to be impressed by rote statements reflecting, to my mind, a total lack of attention or at least a very lazy selective attention to statements from equally lazy and equally misinformed people, often journalists or "activists" of a type that have never passed a college level physical science course. Ignorance, we know, runs in circles, scientific and engineering ignorance as well as political ignorance.
I have been opening science books and reading scientific papers for the bulk of my adult life. A huge percentage of them are about waste and so called "waste." Perhaps, I'm the "nobody" about whom these people speak, since I know perfectly well what to do with so called "nuclear waste." It contains, I'm convinced, enough plutonium (as well as americium and neptunium) to save the Greenland Ice Sheet, and in fact, the world.
Tears in Rain.
I wish you a safe and pleasant weekend.
Profile Information
Gender: MaleCurrent location: New Jersey
Member since: 2002
Number of posts: 33,538